
November 2, 2015
By testing over 4,000 genes in human tumors, a Salk team uncovered an enzyme responsible for suppressing a common and deadly lung cancer
By testing over 4,000 genes in human tumors, a Salk team uncovered an enzyme responsible for suppressing a common and deadly lung cancer
LA JOLLA–Scientists at the Salk Institute have uncovered a molecule whose mutation leads to the aggressive growth of a common and deadly type of lung cancer in humans.
This enzyme, called EphA2, normally polices a gene responsible for tissue growth. But when EphA2 is mutated, the Salk team discovered, cellular systems can run amok and quickly develop tumors. The new work, published the week of November 2, 2015 in PNAS, suggests that EphA2 could be a new target for a subset of lung cancer, which affects nonsmokers as well as smokers, and is the leading cause of cancer-related deaths worldwide.
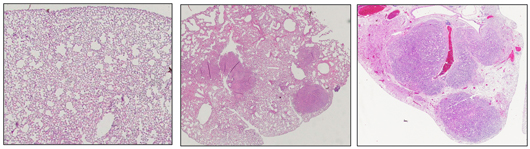
Salk scientists discover a molecule in humans called EphA2 that acts as a tumor suppressor in certain types of lung cancer. This image shows normal mouse lung tissue (left) and lungs with tumors resulting from a KRAS mutation (center). When EphA2 is knocked out, the KRAS mutation results in many more and larger tumors during the same time frame (right).
Click here for a high-resolution image.
Image: Courtesy of the Salk Institute for Biological Studies
“Sometimes there are hundreds of mutations in the genes of a patient’s tumors, but you don’t know whether they are drivers of the disease or byproducts,” says senior author Inder Verma, professor of genetics and holder of Salk’s Irwin and Joan Jacobs Chair in Exemplary Life Science. “We found a new way by which to identify cancer suppressor genes and understand how they could be targeted for therapies.”
Two gene mutations in particular are known to spur the growth of human tumors: KRAS and p53. Though both genes have been heavily studied, they are difficult to therapeutically target, so the Salk team decided to look at genes that might police KRAS and p53 instead.
The researchers narrowed in on the 4,700 genes in the human genome related to cellular signaling–specifically, genes that have the ability to tamp down cell growth and proliferation. Then the team adapted a genetic screening technique to quickly and efficiently test the effect of these thousands of genes on tumor development. In animal models, the Salk team found that 16 of these cell-signaling genes produced molecules that had a significant effect on KRAS- and p53-related tumors.
Of these 16 molecules, one especially stood out: the EphA2 enzyme, originally discovered in the lab of another Salk scientist, Tony Hunter. Previously, EphA2’s significance in lung cancer was unclear, but the team discovered that its absence let KRAS-associated tumors grow much more aggressively.
“With a mutation in KRAS, a tumor forms in 300 days. But without EphA2, the KRAS mutation leads to tumors in half the time, 120 to 150 days,” says Verma, who is also an American Cancer Society Professor of Molecular Biology. “This molecule EphA2 is having a huge effect on restraining cancer growth when KRAS is mutated.” Mutated KRAS is a common culprit in approximately 10 to 20 percent of all cancers, particularly colon cancer and human lung cancer.

From left: Salk researchers Yifeng Xia, Eugene Ke, Narayana Yeddula and Inder Verma
Click here for a high-resolution image.
Image: Courtesy of the Salk Institute for Biological Studies
“Since activating EphA2 led to the suppression of both cell signaling and cell proliferation, we believe that the enzyme might serve as a potential drug target in KRAS-dependent lung adenocarcinoma,” says Narayana Yeddula, a Salk research associate and first author of the paper.
A 10-year national project called the Cancer Genome Atlas mapped the genomes of hundreds of patients for over 20 different cancers and uncovered a number of related genetic mutations, though the role of these mutations has not been well understood in lung cancer (especially adenocarcinoma, which makes up almost a quarter of all lung cancers). From the Cancer Genome Atlas data, the Salk team found that genetic alterations of EphA2 were detected in 54 out of 230 patients with adenocarcinoma. The team also found, surprisingly, that the loss of EphA2 activated a pathway commonly associated with cancer (dubbed Hedgehog) that promotes tumor growth.
“Oddly, among human lung cancer patients with EphA2 mutations, around 8 percent of patients actually have high EphA2 expression. So, in some instances, EphA2 is not suppressing tumors and may be context-dependent. Therefore, we need to carefully evaluate the molecule’s function when designing new therapeutics,” adds Yifeng Xia, a Salk staff researcher involved in the work.
Other authors on the paper were Eugene Ke of the Salk Institute and Joep Beumer of the Salk Institute and the Hubrecht Institute in the Netherlands.
This work was supported in part by an NIH grant, a Salk Cancer Center Core grant, Ipsen, the H.N. and Frances C. Berger Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust.
JOURNAL
PNAS
TITLE
Screening for Tumor Suppressors: Loss of ephrin receptor A2 cooperates with oncogenic KRAS in promoting lung adenocarcinoma
AUTHORS
Narayana Yeddula, Yifeng Xia, Eugene Ke, Joep Beumer, and Inder M. Verma
Office of Communications
Tel: (858) 453-4100
press@salk.edu
Unlocking the secrets of life itself is the driving force behind the Salk Institute. Our team of world-class, award-winning scientists pushes the boundaries of knowledge in areas such as neuroscience, cancer research, aging, immunobiology, plant biology, computational biology and more. Founded by Jonas Salk, developer of the first safe and effective polio vaccine, the Institute is an independent, nonprofit research organization and architectural landmark: small by choice, intimate by nature, and fearless in the face of any challenge.