
April 21, 2009
Neuronal growth factor receptor protects the sympathetic nervous system in Alzheimer's disease
Neuronal growth factor receptor protects the sympathetic nervous system in Alzheimer's disease
La Jolla, CA—New research at the Salk Institute for Biological Studies casts the role of a neuronal growth factor receptor—long suspected to facilitate the toxic effects of beta amyloid in Alzheimer’s disease—in a new light, suggesting the molecule actually protects the neuron in the periphery from beta amyloid-induced damage.
The receptor molecule in question, a protein better known as p75, regulates neuronal growth, survival, and degeneration, and guides nerve fibers in growing embryos to their final destinations. Some studies have suggested that it also exacerbates the neurotoxicity associated with beta amyloid deposits, which litter the brains of Alzheimer’s patients, giving the molecule its questionable reputation.
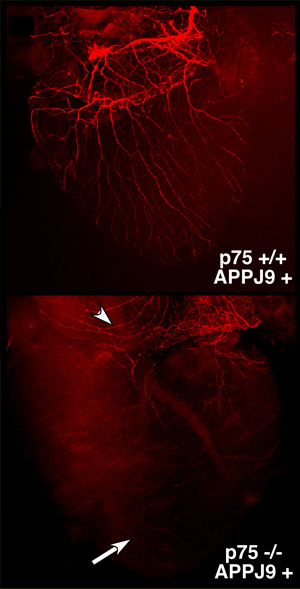
Top: The neuronal wiring (shown in red) of the heart in Alzheimer’s mice appears normal. Bottom: The sympathetic innervation in p75-deficient Alzheimer’s mice is severely impaired.
Image: Courtesy of Lee Laboratory, Salk Institute for Biological Studies.
Yet a team of scientists in the laboratory of Kuo-Fen Lee, Ph.D., a professor in the Clayton Foundation Laboratories for Peptide Biology, found that p75 instead has a neuroprotective effect on the sympathetic nervous system in mice that were genetically engineered to develop Alzheimer’s disease.
Their findings, published in this week’s early online edition of the Proceedings of the National Academy of Sciences, challenge the prevailing view of p75’s harmful role in the condition and could lead to new insights and, ultimately, new protocols for managing the secondary deficits that accompany dementia and memory loss in Alzheimer’s.
Scientific interest in the peripheral nervous system has been growing as investigators studying neurodegenerative diseases seek new insights into disease progression. “How a disease damages the peripheral nervous system could add a great deal to our understanding of its process, possibly leading to applications down the line that impact patient management and quality of life issues,” says Lee, who led the study.
Proteins, like people, are often judged by the company they keep. p75, for instance, belongs to the same family as tumor necrosis factor and was widely thought to mediate cell death in some context. Various in vitro studies have examined p75 in combination with beta amyloid, seeking evidence that it helps induce nerve cell death in Alzheimer’s disease.
To gather evidence about p75 and the sympathetic nervous system, Lee’s team crossed a mouse model for Alzheimer’s disease with a line of mice genetically modified to lack the gene for p75. Without p75, they theorized, the neurotoxic effects of beta amyloid would be reduced, and the mice would show fewer Alzheimer’s symptoms.
“The role of p75 had been controversial for some time, but based on the evidence at the time, we expected to see indications that it mediates beta amyloid neurotoxicity,” says co-first author Tasha Bengoechea, Ph.D., a former graduate student in Lee’s lab. “We thought removing p75 while overexpressing amyloid would have a positive effect on neuron viability. The opposite was true.”
Along with profound motor problems, the p75-deficient mice exhibited severe defects in the wiring of nerves to multiple organs, and the majority died within just three weeks. (Mice normally live up to two years.) When the researchers scaled down the production of toxic beta amyloid by deleting one copy of BACE1, which encodes the molecular shears that make the first cut in the production of beta amyloid fragments, the nerves in the sympathetic nervous system of p75-deficient mice were substantially restored.
“This is the first time the interplay between p75 and beta amyloid in the peripheral sympathetic system, a system that has not been paid much attention before, has been demonstrated,” adds postdoctoral researcher and co-first author Zhijiang Chen, Ph.D. “Our findings will ultimately help to design novel strategies to treat the symptoms of the Alzheimer’s disease and improve the quality of life for Alzheimer’s disease patients.”
The study was funded by a grant from the National Institutes of Health. Researchers who also contributed to the work included postdoctoral researcher Deborah O’Leary, Ph.D., of the Salk Institute’s Clayton Foundation Peptide Biology Laboratory, and Eliezer Masliah, Ph.D., a professor in the Department of Neurosciences at the University of California, San Diego.
###
About the Salk Institute for Biological Studies
The Salk Institute for Biological Studies is one of the world’s preeminent basic research institutions, where internationally renowned faculty probe fundamental life science questions in a unique, collaborative, and creative environment. Focused on both discovery and mentoring future generations of researchers, Salk scientists make groundbreaking contributions to our understanding of cancer, aging, Alzheimer’s, diabetes, and cardiovascular disorders by studying neuroscience, genetics, cell and plant biology, and related disciplines.
Faculty achievements have been recognized with numerous honors, including Nobel Prizes and memberships in the National Academy of Sciences. Founded in 1960 by polio vaccine pioneer Jonas Salk, M.D., the Institute is an independent nonprofit organization and architectural landmark.
Office of Communications
Tel: (858) 453-4100
press@salk.edu