
May 19, 2016
New findings yield insights into how plants get their traits
New findings yield insights into how plants get their traits
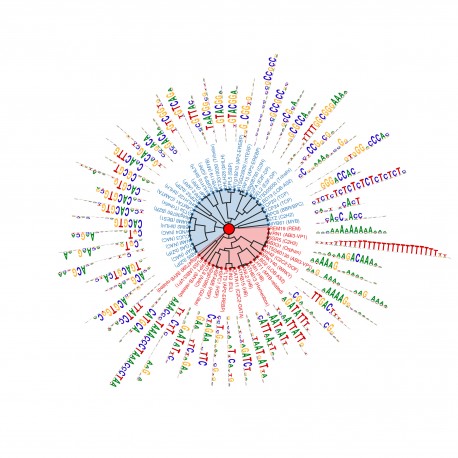
LA JOLLA—A new technique developed by Salk Institute scientists for rapidly mapping regions of DNA targeted by regulatory proteins could give scientists insight into what makes some plants drought tolerant or disease resistant, among other traits.
Revealing this landscape of protein-binding zones on DNA, collectively dubbed the “cistrome,” shows how plants control where and when genes are expressed. Previous methods for mapping the cistrome in plant cells were difficult and slow, but the new approach, detailed in the May 19, 2016 issue of Cell, overcomes those hurdles to offer a sweeping view of this critical aspect of genetic regulation.
Click here for a high-resolution image
Credit: Salk Institute
“This is one of the first efforts to globally characterize all the regulatory elements in a plant genome,” says senior author Joseph Ecker, professor and director of Salk’s Genomic Analysis Laboratory and holder of the Salk International Council Chair in Genetics. “The cistrome has been a missing piece of information for trying to understand how plants function.”
Lots of information in plant and animal cells is contained in “coding” stretches of DNA that have the instructions to make proteins, the physical workhorses of cells. But researchers are increasingly realizing that other sections of the genome have elements that control when and how cells make these proteins. Among these “non-coding” bits of DNA are spots where proteins called transcription factors bind to manage the activation of neighboring coding genes.
“Many studies in humans and animal models show that non-coding changes are really important for understanding hereditary disorders and diseases like cancer,” says Shao-shan Carol Huang, a Salk research associate and co-first author of the paper. “Being able to figure out what these non-coding regions are doing in plants is equally critical.”
In the past, scientists could determine where just one or two transcription factors at a time were attaching to plant genomes, but the experiments were slow going. Ecker and his colleagues wanted to fully map where the hundreds—or even thousands—of known transcription factors bind, so they needed a faster way to map the sites.
To achieve this cistrome mapping, the researchers created a system where they could add a tagged transcription factor to a library of DNA, let it bind and then isolate all the DNA-protein pairs. The method, called DNA affinity purification sequencing (DAP-seq), vastly expands how much more information scientists can glean about transcription factors and their binding sites.

Click here for a high-resolution image
Credit: Salk Institute
“Our system is inexpensive and scalable,” says Ronan O’Malley, a staff scientist in the Ecker lab and co-first author of the paper. “It allows us to capture the full collection of transcription binding sites so we have a complete codebook.” Moreover, he said, it doesn’t require sophisticated or specialized laboratory equipment that most plant labs wouldn’t already have access to.
To test out the utility of DAP-seq, Ecker, Huang, O’Malley and their colleagues mapped where 529 transcription factors bound to the genome of Arabidopsis thaliana, the plant most studied by scientists. They identified 2.7 million binding sites. They then repeated the experiments using DNA containing or not containing cytosine methylation—a process of marking the genome’s surface with chemical methyl tags to further inhibit or activate genes. The binding patterns of about three-quarters of the transcription factors that they tested changed.
“This allowed us to expose binding sites that we might miss if we didn’t remove the methylation. With this approach we can see binding sites that may be active in only a subset of cells or tissues,” says O’Malley. The new results show not only how regulatory proteins alter gene expression, but the roles the epigenomic methylation marks may play in this regulation.
In a separate paper, published May 2016 in Nature Plants, Ecker and collaborating groups at Duke University and the University of Western Australia revealed that different types of cells in the Arabidopsis root have different patterns of methylation. Using DAP-seq, they’ll now be able to study how those patterns in root cells affect the binding of transcription factors. In addition, they would like to study how different transcription factors interact with each other, and try applying the method to other plant species and to human cells.
“The beauty of this is that it can be done in any plant, many of which don’t have the tools available in Arabidopsis,” says Ecker, who is also a Howard Hughes Medical Institute and a Gordon and Betty Moore Foundation investigator. “And this can give us a window into how genetic or epigenetic variations in regulatory sequences affect their traits.”
Other researchers on the study were Liang Song, Mathew G. Lewsey, Anna Bartlett and Joseph R. Nery, all of the Salk Institute; and Mary Galli and Andrea Gallavotti of Rutgers University.
The work and the researchers involved were supported by grants from the National Science Foundation, the Gordon and Betty Moore Foundation and the Howard Hughes Medical Institute.
JOURNAL
Cell
AUTHORS
Ronan C. O’Malley, Shao-shan Carol Huang, Liang Song, Mathew G. Lewsey, Anna Bartlett, Joseph R. Nery, and Joseph R. Ecker of the Salk Institute; Mary Galli and Andrea Gallavotti of Rutgers University.
Office of Communications
Tel: (858) 453-4100
press@salk.edu
Unlocking the secrets of life itself is the driving force behind the Salk Institute. Our team of world-class, award-winning scientists pushes the boundaries of knowledge in areas such as neuroscience, cancer research, aging, immunobiology, plant biology, computational biology and more. Founded by Jonas Salk, developer of the first safe and effective polio vaccine, the Institute is an independent, nonprofit research organization and architectural landmark: small by choice, intimate by nature, and fearless in the face of any challenge.