
January 20, 2010
LA JOLLA, CA—A viral infection is like an uninvited, tenacious houseguest in the cell, using a range of tricks to prevent its eviction. Researchers at the Salk Institute for Biological Studies have identified one of the key proteins allowing herpes simplex virus (HSV) DNA to fly under the radar of their hosts’ involuntary hospitality.
Their findings, to be published in a forthcoming issue of The EMBO Journal, reveal one of the tactics that HSV employs in order to overcome its hosts’ defenses and may suggest a common mechanism by which viruses can successfully infect host cells.
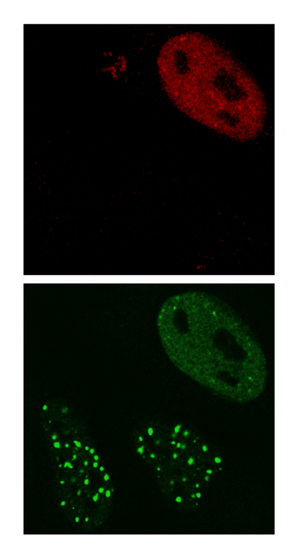
When cells with DNA damage are infected with HSV-1 virus (top image, virus shown in red) the viral ICP0 protein prevents the DNA repair proteins (bottom image, DNA repair proteins shown in green) from accumulating at sites of DNA damage.
Image: Courtesy of Dr. Caroline Lilley, Salk Institute for Biological Studies
HSV, like all viruses, requires a living host in order to multiply. But before it can hijack the cellular machinery to produce scores of copies of itself, it needs to evade the cell’s security system. “We found that detection of the viral DNA by the host cell is an important barrier that the virus must overcome in order to achieve its goal,” says Matthew Weitzman, Ph.D., associate professor in the Laboratory of Genetics, who led the study. “For this purpose, it brings along a protein that shuts down the normal cellular responses that would otherwise recognize and silence it.”
To the host cell, invading viral DNA looks just like the product of DNA damage, which must be repaired or removed in order for the cell to stay healthy. As a result, DNA “security guards” continuously patrol our cells looking for unusual bits of DNA. “We reasoned that viral DNA would be recognized by the cell’s DNA repair machinery and that the virus must somehow manipulate the cell’s response to this foreign DNA,” explains Weitzman.
To test this hypothesis, Weitzman and his team looked at what happens in a virus-infected cell when its DNA is damaged. In a normal cell, DNA damage sensor proteins rush to the site of damage. In cells infected with HSV, however, the cells’ emergency repair teams don’t respond correctly. “The virus effectively overrides the cell’s DNA damage response in order to prevent its own DNA from being recognized,” says Weitzman.
The team went on to identify a single viral protein that is to blame for knocking out the cell’s security system, a protein called ICP0. They discovered that it flags for destruction two important DNA “security guards,” the proteins called RNF8 and RNF168, thereby taking out the DNA damage response in human cells in one big swipe.
ICP0 attaches so-called ubiquitin marks, which instruct the cell to get rid of the very proteins that protect it. With RNF8 and RNF168 safely out of the way, the virus can begin to take over.
Delving deeper, the team looked at the role of these DNA “security guards” that are singled out by ICP0. Surprisingly, RNF8 and RNF168 also leave ubiquitin tags, but in this case, they mark regions of damage. They tag a protein called histone H2A, which directs DNA damage response proteins to accumulate at the sites of damage. The Salk team discovered that by removing RNF8 and RNF168, the viral ICP0 protein results in a decrease to the tag on the cellular H2A protein.
“We found that HSV targets the mark that is required to keep DNA damage sensors at damage sites,” says postdoctoral researcher and first author Caroline Lilley, Ph.D. “We now think that HSV deliberately removes this mark so that the virus can infect cells without any trouble from its new host.”

Mira Chaurushiya, Matt Weitzman and Caroline Lilley.
The findings highlight the importance of these histone marks in DNA damage. “By identifying how HSV dismantles the host’s defense systems, we are shown the key steps, not only in viral infections, but also in the human DNA damage response,” Weitzman explains.
HSV may have evolved this weapon because our cells use histone ubiquitination to try to silence gene expression from the viral DNA. “Ubiquitination would be a great way for the cell to silence the viral genome, and ICP0 provides the counterattack by the virus, so the virus and the cell are battling it out at that point,” says Mira Chaurushiya, a graduate student in the lab and contributor to the study.
This work may point to a general mechanism viruses use to overcome the cell’s defense. “DNA damage signaling and ubiquitination may be part of an anti-viral defense mechanism,” explains Lilley. “Part of the cell’s defense is to try to silence viral genomes, and viral proteins have to prevent this in order to achieve infection.”
Along with Weitzman, Lilley and Chaurushiya, other contributors to this work were Sebastien Landry and Junghae Suh from the Salk Institute’s Laboratory of Genetics (J.S. is now at Rice University, Houston); Stephanie Panier and Daniel Durocher from the Samuel Lunenfeld Research Institute, Mount Sinai Hospital in Toronto, Canada; Chris Boutell and Roger D. Everett at the University of Glasgow, UK; and Grant S. Stewart at Birmingham University, UK.
-Claire Attwooll
About the Salk Institute for Biological Studies
The Salk Institute for Biological Studies is one of the world’s preeminent basic research institutions, where internationally renowned faculty probe fundamental life science questions in a unique, collaborative, and creative environment. Focused both on discovery and on mentoring future generations of researchers, Salk scientists make groundbreaking contributions to our understanding of cancer, aging, Alzheimer’s, diabetes, and cardiovascular disorders by studying neuroscience, genetics, cell and plant biology, and related disciplines.
Faculty achievements have been recognized with numerous honors, including Nobel Prizes and memberships in the National Academy of Sciences. Founded in 1960 by polio vaccine pioneer Jonas Salk, M.D., the Institute is an independent nonprofit organization and architectural landmark.
Office of Communications
Tel: (858) 453-4100
press@salk.edu